FDA AI Medical Device Approval: 2026 Fast-Track Guide

The landscape of healthcare is undergoing a profound transformation, driven by the rapid advancements in Artificial Intelligence (AI). From diagnostic imaging to personalized treatment plans, AI in medical devices promises to revolutionize patient care, enhance efficiency, and unlock new frontiers in medical discovery. However, bringing these innovative technologies to market requires navigating a complex regulatory environment, with the U.S. Food and Drug Administration (FDA) at the forefront. As we approach 2026, the FDA is refining its approach, particularly with updated 90-day fast-track guidelines designed to accelerate the approval of groundbreaking AI medical devices.
Understanding the nuances of these guidelines is paramount for developers, manufacturers, and stakeholders in the healthcare technology sector. This comprehensive guide delves into the intricacies of the FDA’s regulatory framework for AI in medical devices, with a specific focus on the highly anticipated 2026 updates to the 90-day fast-track program. Our aim is to provide clarity on the pathways to approval, highlight key considerations for compliance, and offer strategic insights for successfully bringing your AI-powered innovations to patients.
The Evolving Role of AI in Medical Devices
AI’s integration into medical devices is not merely an incremental improvement; it represents a paradigm shift. These devices, ranging from software as a medical device (SaMD) to hardware integrated with sophisticated algorithms, can analyze vast datasets, identify subtle patterns, and assist clinicians in making more informed decisions. Examples include AI-powered algorithms for detecting early signs of disease from medical images, predictive analytics for patient deterioration, and adaptive treatment systems that personalize therapy based on real-time data.
The potential benefits are immense: earlier diagnoses, more effective treatments, reduced healthcare costs, and improved patient outcomes. However, the unique characteristics of AI – particularly its adaptive and learning capabilities – present novel challenges for regulatory oversight. Unlike traditional medical devices with fixed functionalities, many AI algorithms can continuously learn and evolve, raising questions about ongoing safety, efficacy, and transparency.
FDA’s Regulatory Framework for AI Medical Devices: A Historical Perspective
The FDA has been proactive in addressing the unique challenges posed by AI in medical devices. Recognizing the rapid pace of innovation, the agency has sought to develop a regulatory framework that fosters technological advancement while safeguarding public health. Historically, medical devices have been categorized into three classes (Class I, II, and III) based on risk, with corresponding premarket submission pathways: 510(k) premarket notification, De Novo classification request, and Premarket Approval (PMA).
For AI, the FDA initially adapted existing frameworks, but soon realized the need for more tailored guidance. Key milestones include the release of discussion papers, guidance documents on SaMD, and the establishment of the Digital Health Center of Excellence (DHCoE). These initiatives underscored the FDA’s commitment to creating a predictable and efficient regulatory path for AI medical devices. The focus has been on ensuring that AI algorithms are well-validated, perform reliably, and are transparent in their intended use and limitations.
The Need for a Fast-Track: Accelerating Innovation Safely
The traditional regulatory pathways, while robust, can sometimes be lengthy, potentially delaying patient access to critical innovations. For AI-driven technologies that offer significant advantages over existing methods or address unmet medical needs, a faster route to market can be crucial. This is where fast-track programs come into play. The FDA has long utilized mechanisms like Breakthrough Device Designation to expedite the development and review of certain medical devices that provide more effective treatment or diagnosis of life-threatening or irreversibly debilitating diseases or conditions.
The 2026 updates to the 90-day fast-track guidelines for AI in medical devices build upon this philosophy. The goal is to create a more streamlined and predictable pathway for specific types of AI innovations, ensuring that promising technologies reach patients sooner without compromising safety or effectiveness. This initiative reflects the FDA’s understanding that the benefits of certain AI-powered devices warrant an accelerated review, provided stringent criteria are met.
Decoding the 2026 Updated 90-Day Fast-Track Guidelines for FDA AI Medical Devices
The 2026 updates represent a significant refinement of the FDA’s approach to expedited review for AI in medical devices. While the full details are still being finalized and will be subject to public comment, key themes and anticipated changes are emerging. These updates are designed to clarify the eligibility criteria, streamline the submission process, and enhance predictability for developers seeking accelerated approval.
Eligibility Criteria for the 90-Day Fast-Track
Not all AI medical devices will qualify for the 90-day fast-track. The FDA is expected to focus on devices that:
- Address Unmet Medical Needs: The AI device must target a condition or disease for which no satisfactory alternative treatment or diagnosis exists, or it must offer a significant advantage over existing options.
- Demonstrate Substantial Clinical Benefit: Preliminary evidence must suggest that the device provides a clinically meaningful advantage in diagnosis, treatment, or patient management. This could include improved accuracy, earlier detection, or better patient outcomes.
- Employ Well-Characterized AI: The underlying AI algorithms must be sufficiently characterized, with clear documentation of their development, training data, validation methods, and performance metrics. Transparency and explainability of the AI model will be crucial.
- Manage Risks Effectively: Developers must demonstrate a robust risk management plan, addressing potential biases in AI, cybersecurity vulnerabilities, and strategies for monitoring post-market performance.
- Adaptive AI Considerations: For continuously learning AI models, the FDA will likely require a predetermined change control plan (PCCP) outlining how modifications will be managed and validated without requiring a new premarket submission for every update.
Streamlined Submission Process
The 90-day fast-track is not just about a shorter review period; it also implies a more efficient submission and interaction process. Developers can expect:
- Early Engagement with FDA: Increased opportunities for pre-submission meetings and discussions with FDA experts to clarify requirements and address potential issues early in the development cycle.
- Tailored Data Requirements: The FDA may provide more specific guidance on the types and formats of data required for AI devices, potentially focusing on real-world evidence (RWE) and real-world data (RWD) where appropriate.
- Iterative Review: A more collaborative and iterative review process, allowing for quicker feedback and resolution of deficiencies.
- Clear Communication Pathways: Enhanced communication channels between the FDA and applicants to ensure transparency and efficiency throughout the review.
Strategic Considerations for Developers of FDA AI Medical Devices
For companies aiming to leverage the 90-day fast-track for their AI medical devices, a proactive and strategic approach is essential. Here are key areas to focus on:
1. Robust Data Strategy and Management
The performance and reliability of any AI algorithm are intrinsically linked to the quality and representativeness of its training data. Developers must prioritize:
- Data Sourcing and Curation: Ensuring that data is ethically acquired, diverse, and free from biases that could lead to discriminatory or inaccurate AI performance.
- Data Annotation and Validation: Implementing rigorous processes for data annotation by qualified experts and thorough validation of the data’s integrity.
- Data Governance: Establishing clear policies for data privacy, security, and traceability throughout the device lifecycle.
2. Comprehensive Algorithm Validation and Performance Evaluation
Beyond traditional software validation, AI algorithms require specific validation methodologies. This includes:
- Technical Validation: Assessing the algorithm’s accuracy, precision, recall, and other relevant metrics using independent test datasets.
- Clinical Validation: Demonstrating the device’s performance in real-world clinical settings, ideally through prospective studies.
- Bias Detection and Mitigation: Actively identifying and addressing potential biases in the algorithm’s output, particularly concerning different demographic groups.
- Explainability and Interpretability: Where feasible and clinically relevant, developing methods to explain how the AI arrived at its conclusions to foster trust and facilitate clinical adoption.
3. Quality Management System (QMS) Tailored for AI
An effective QMS is foundational for any medical device. For AI medical devices, the QMS needs to specifically address:
- Software Development Life Cycle (SDLC) for AI: Integrating AI-specific development practices, including version control for models and datasets, and robust testing protocols.
- Risk Management: Identifying and mitigating risks unique to AI, such as adversarial attacks, data drift, and performance degradation over time.
- Post-Market Surveillance for Adaptive AI: Establishing systems for continuous monitoring of AI performance in the field, detecting performance shifts, and managing updates or retraining in accordance with the PCCP.
4. Cybersecurity and Data Privacy
AI medical devices often handle sensitive patient data and can be vulnerable to cyber threats. Robust cybersecurity measures are non-negotiable:
- Security by Design: Integrating security considerations throughout the design and development process.
- Vulnerability Management: Regularly assessing and patching vulnerabilities to protect against unauthorized access or manipulation.
- Data Encryption and Access Controls: Implementing strong encryption for data at rest and in transit, along with stringent access controls.
5. Human Factors and Usability
Even the most advanced AI medical device will fail if it’s not intuitive and safe for healthcare professionals to use. Human factors engineering is crucial to:
- Optimize User Interface: Designing interfaces that clearly present AI outputs and insights, minimizing cognitive load and potential for error.
- Address Alert Fatigue: Ensuring that AI-generated alerts are actionable and effectively prioritized to prevent clinicians from becoming overwhelmed.
- Training and Education: Providing comprehensive training materials to help users understand the device’s capabilities, limitations, and appropriate use.
The Role of Predetermined Change Control Plans (PCCP) in Adaptive AI
One of the most significant advancements in the FDA’s thinking for AI medical devices, particularly those with adaptive or continuously learning algorithms, is the emphasis on Predetermined Change Control Plans (PCCP). A PCCP is a framework submitted to the FDA that outlines the types of modifications an AI algorithm can undergo after initial market authorization without requiring a new premarket submission for each change.
The PCCP typically specifies:
- Types of Modifications: What kinds of changes are anticipated (e.g., updates to training data, adjustments to algorithm parameters).
- Methods for Validation: How each type of change will be validated to ensure continued safety and effectiveness.
- Performance Monitoring: The metrics and procedures for continuously monitoring the AI’s performance in the real world.
- Transparency and Documentation: How changes will be documented and communicated to users.
For AI medical devices seeking the 90-day fast-track, a well-structured and comprehensive PCCP will likely be a critical component of the submission, demonstrating the developer’s foresight and commitment to managing the evolving nature of their AI product responsibly.
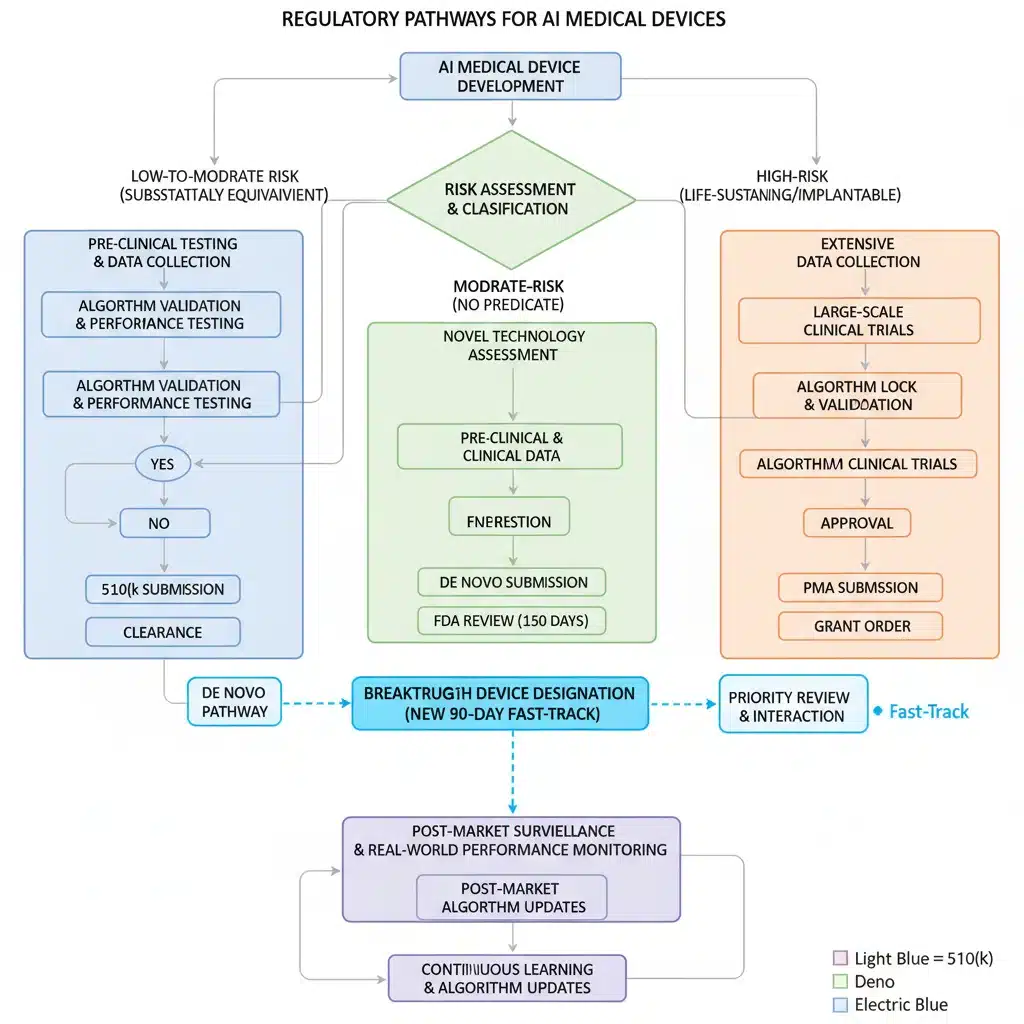
Navigating the Regulatory Pathways: 510(k), De Novo, and PMA for FDA AI Medical Devices
While the 90-day fast-track offers an expedited route, it’s important to understand how it interfaces with the existing regulatory pathways:
- 510(k) Premarket Notification: This pathway is for Class II devices that are substantially equivalent to a legally marketed predicate device. Many AI medical devices, particularly those for diagnostic support, may fall into this category. The fast-track could potentially accelerate the 510(k) review for eligible AI devices.
- De Novo Classification Request: This pathway is for novel Class I or Class II devices for which no predicate exists. AI devices that introduce entirely new functionalities or technologies often go through the De Novo pathway. The fast-track could be particularly valuable here, as De Novo reviews can be complex and time-consuming.
- Premarket Approval (PMA): This is the most stringent pathway, reserved for high-risk Class III devices. While fewer AI devices might fall directly into PMA, those with highly critical functions or significant potential for harm would require this level of scrutiny. The fast-track might offer some efficiencies even within the PMA framework for truly breakthrough AI.
The 90-day fast-track is not a replacement for these pathways but rather an overlay that can expedite the review process within them for qualified AI medical devices. Developers must first determine the appropriate primary regulatory pathway before considering their eligibility for the fast-track program.

Ethical Considerations and Responsible AI Development
Beyond regulatory compliance, the ethical implications of AI in medical devices are paramount. The FDA’s guidelines, particularly for the fast-track, will inherently encourage responsible AI development. Key ethical considerations include:
- Bias and Fairness: Ensuring AI algorithms do not perpetuate or amplify existing health disparities due to biased training data.
- Transparency and Explainability: Providing sufficient insight into how AI makes decisions, especially in critical diagnostic or treatment recommendations.
- Accountability: Clearly defining who is responsible when an AI-powered device makes an error.
- Privacy and Security: Robust protection of patient data used by AI systems.
Developers seeking fast-track approval will need to demonstrate not only technical prowess but also a deep commitment to ethical AI principles, integrating them into every stage of development and deployment.
The Future of FDA AI Medical Devices and the 90-Day Fast-Track
The 2026 updates to the FDA’s 90-day fast-track guidelines for AI in medical devices signal a maturing regulatory landscape that is adapting to technological innovation. This initiative is a clear indication that the FDA is committed to fostering the development of safe and effective AI-powered healthcare solutions while ensuring timely patient access.
As AI continues to evolve, so too will the regulatory framework. Developers should remain agile, staying abreast of the latest guidance and engaging proactively with the FDA. Success in this rapidly changing environment will depend on a combination of scientific rigor, robust quality systems, a deep understanding of regulatory requirements, and an unwavering commitment to ethical principles.
The promise of AI in medicine is immense, offering the potential to transform healthcare as we know it. By strategically navigating the FDA’s updated 90-day fast-track guidelines, innovators have a unique opportunity to bring their life-changing AI medical devices to patients faster, ushering in a new era of intelligent healthcare.
Conclusion
The 2026 updated 90-day fast-track guidelines from the FDA for AI in medical devices represent a pivotal moment for healthcare innovation. These guidelines are designed to expedite the review of groundbreaking AI technologies that address unmet medical needs and demonstrate substantial clinical benefit. For developers, understanding and strategically preparing for these changes is crucial. This includes developing robust data strategies, implementing comprehensive algorithm validation, establishing AI-specific quality management systems, prioritizing cybersecurity and human factors, and integrating ethical considerations throughout the development lifecycle. By embracing these principles and engaging proactively with the FDA, companies can significantly increase their chances of navigating the regulatory landscape successfully and bringing transformative AI medical devices to market swiftly and safely. The future of healthcare is intelligent, and the FDA’s updated fast-track is poised to accelerate its arrival.





